
THE TRUTH IS ALWAYS ON THE OTHER SIDE!
THE TRUTH IS ALWAYS ON THE OTHER SIDE!
Nucleic Acid Snippets - Human Haploid

https://docs.google.com/spreadsheets/d/1uNoijuXDg5tzstEuaxtOrYv3fZ0Nyc405_thJY4-BBg/edit?fbclid=IwAR0lkOMjvpfyZKpV3OSGe3EpaLlBSeBf3RyNLz_NPBd18NoGxLdHjTAEGtU#gid=0
All these so called "C-virus sequences/nucleic acid snippets" are present in various parts of our genome - this is from various diseased cells being rendered harmless/mineralized by macrophages and transported out of the body along with rendered harmless toxins by exosomes(either as DNA or as "reverse transcribed" RNA, since we know that exosomes have both!) or EVs

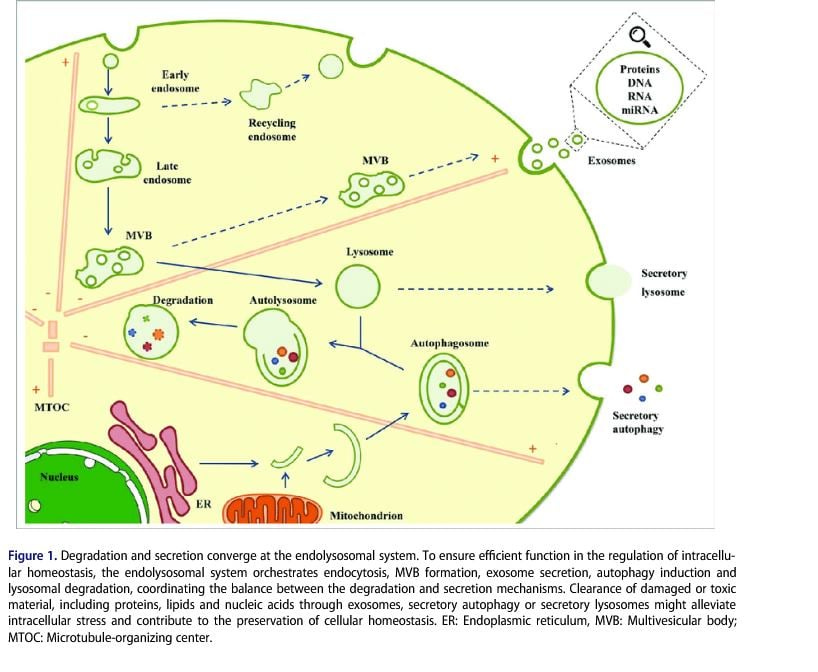
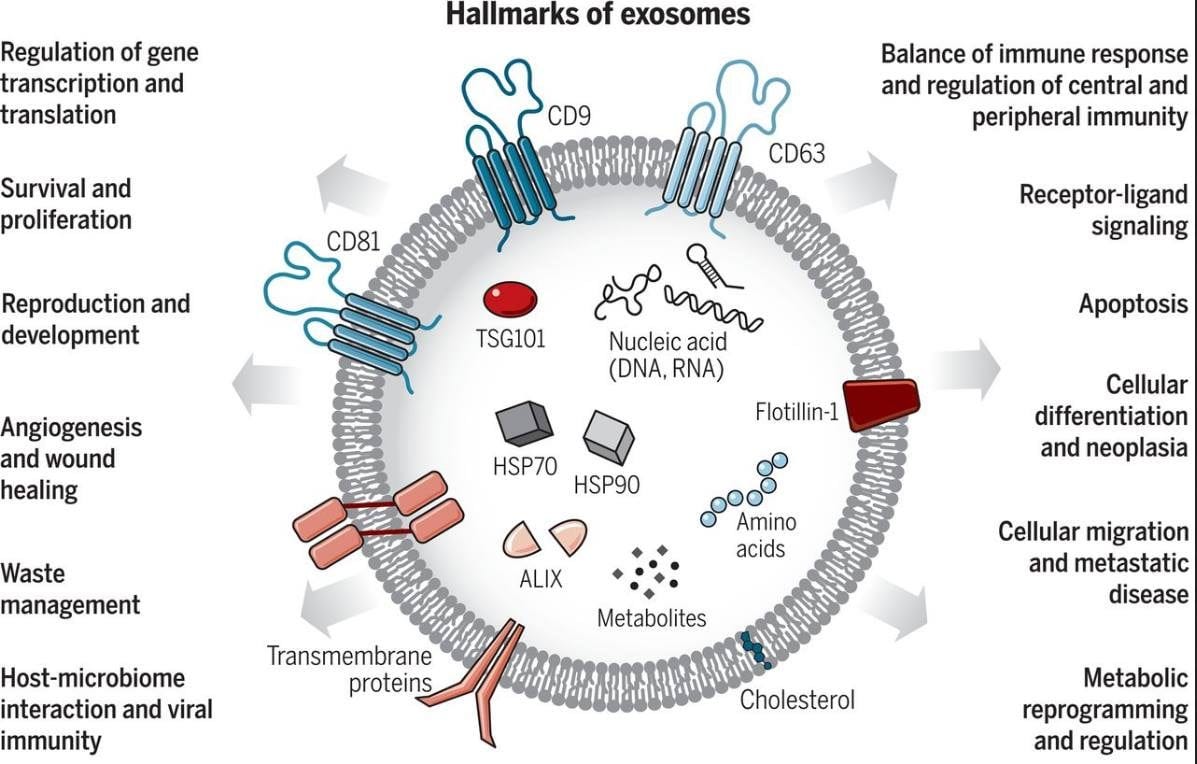
- and THIS has NOTHING to do with a "virus" but is a normal process to keep the body healthy - and confused, ignorant so-called "virologists" pass off bacteriophages that cling to bacterial cell walls as so-called "viruses" and THAT is unscientific and just plain stupid!!!!
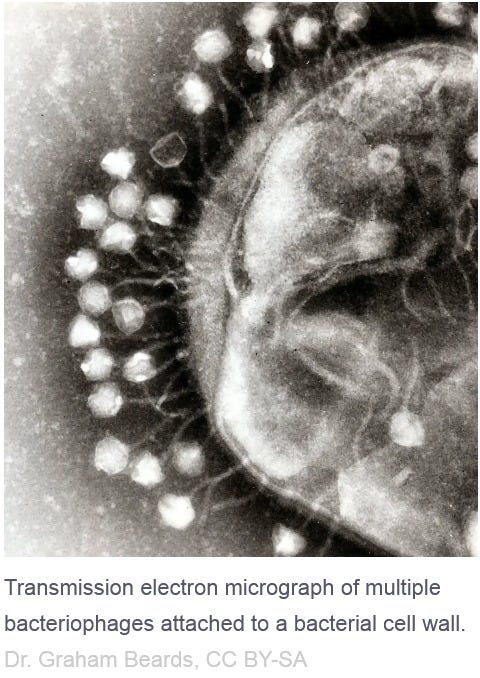
vs.
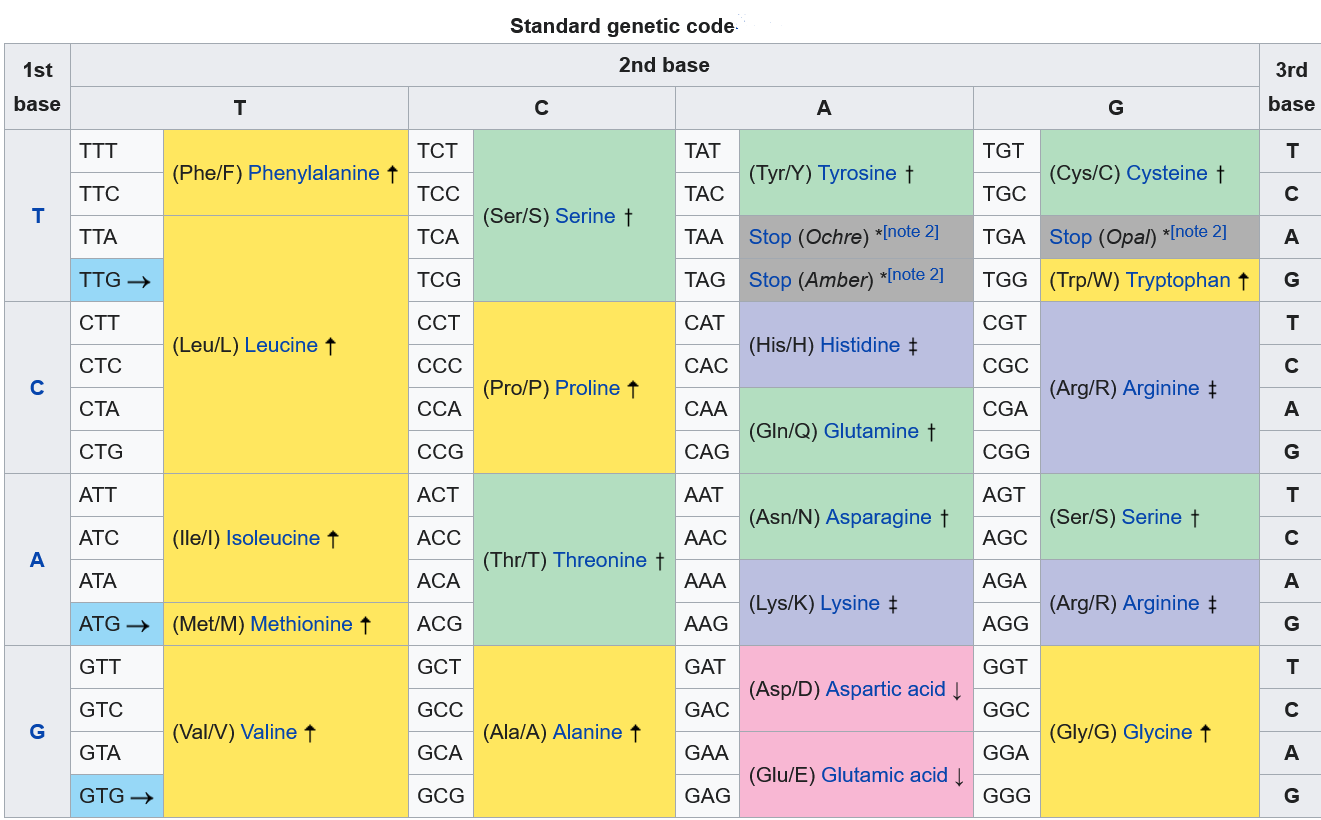
Under the commonly used UPAC system, nucleobases are represented by the first letters of their chemical names guanine, cytosine, adenine, and thymine.
This shorthand also includes eleven "ambiguity" characters associated with every possible combination of the four DNA base and to be able to understand this to some extent, you have to look at this
and internalize it and this https://www.genome.gov/Pages/Research/Sequencing/BACLibrary/HydatidiformMoleBAC021203.pdf (THIS has not been clarified to this day and these gaps still exist). "The central task of human genetics is the correlation of phenotype and genotype. Much of this effort depends on our ability to track unique DNA by association or linkage with phenotype. The revelation that a significant fraction (~5%) of our genome is composed of recent segmental duplications has a serious impact on the work of human geneticist and the final assembly of the human genome (Bailey et al. 2001; Eichler 2001) . Segmental duplications may span large distances of genomic sequence (in some cases 100’s of kb), share a high degree of sequence identity (>99%), can harbor genes, and, unlike, other classes of repetitive sequences can not be distinguished as such, a priori. In essence, these properties have made a portion of our genome intractable by the standard fare of molecular techniques applied within our field. The potential for such regions to rearrange and create structural polymorphisms (Giglio et al. 2001; Giglio et al. 2002; Osborne et al. 2001) has further confounded traditional linkage analysis inthese regions (Nelson Freimer and Leena Peltonen, personal communications). The development of human SNP maps is similarly hampered leading to misleadingly high density of SNPs over duplicated regions-where collapse of SNPs and paralogous sequence variants occur (Bailey et al. 2002; Estivill et al. 2002) . Duplicated segments pose ser ious problems for the assembly and annotation of the human genome. Even among chromosomes that are near completion, there are still large (>100 kb) gaps which will require specialized efforts to fill or regions in which the present assembly is suspect. M any of these gaps lie within highly duplicated regions that are not necessarily refractory to subcloning. Instead, these same regions contain many highly duplicated segments in which the degree of sequence variation among duplicated loci (paralogous sequence variation) approaches levels of allelic variation. Finally, it has become increasingly apparent that the segmental duplications themselves provide the molecular basis for many human genetic disorders, including complex genetic disease traits (Gratacos et al. 2001; Stankiewicz and Lupski 2002). Their biological resolution of these regions is therefore essential for a complete understanding of the genetic basis of the human disease. First and foremost, however, it is essential that such highly paralogous regions be identified, their locations refined and their sequence correctly assembled into the human reference genome. One of the key impediments in resolving the complexity of these regions is the diploid and polymorphic nature of the human genome. In the past, the distinction between allelic versus polymorphic variation has been successfully circumvented by the use of genetic material of haploid complexity. This has included the use of monchromosomal source material such as genomic cosmid libraries and/or monochromosomal somatic cell hybrids (Horvath et al. 2000; Johnson et al. 2001). Such resources , while helpful, are not satisfactory for establishing continuity across large (>100 kb) regions of segmental duplication at a genome-wide level. The final sequence and assembly of the Y chromosome (which is unusually enriched for segmental duplications) was achieved in large part due to the fact that all the “BAC clones [came] from one man’s Y chromosome” (Kuroda-Kawaguchi et al. 2001) . Sequence assembly was therefor e not impaired by polymorphism and all sequence variants represented distinct copies of paralogous sequence."